Chapter 1
Discovering insulin
1.1 Introduction
The disease Diabetes Mellitus was first described in an Egyptian papyrus,
discovered by Ebers in the tomb of Thebes in Egypt in 1862, which is said to
have been written between 3000 and 1500 BC. The first use of the term 'Diabetes
Mellitus' is accredited to Aretaeus of Cappadocia and Apolonius of Memphis in
the second century AD. 'Diabetes' stems from the Greek word for 'pipe-like'
because nutrients begin to pass through the system rather than being utilised.
'Mellitus' is Latin for 'honey' or 'sweet', to distinguish the disease from
'Diabetes Insipidus', which is a pituitary disorder in which large volumes of
sugar-free urine are passed.
In the middle of the nineteenth century, evidence from autopsies started to
suggest a link between the pancreas and Diabetes Mellitus. Diabetics were
sometimes seen to have pancreas damage, and patients with damaged pancreases
almost always had diabetes. In 1869 Langerhans discovered the existence of two
systems of cells in the pancreas: the acinar cells, secreting the pancreatic
juice into the digestive system, and islets floating between the acini, with
some as yet unknown function. In 1889 Minkowski and Von Mering depancreatised a
dog, causing a state of polyuria indistinguishable from diabetes. This was the
first direct evidence of the link between diabetes and the pancreas. They also
showed that it was not the absence of the pancreatic juice that caused diabetes
by studying the effect of ligating the pancreatic ducts rather than removing the
whole pancreas. In most cases this caused minor digestive problems, but never
diabetes.
The Frenchman Hédon proved in 1893 that a total pancreatectomy was necessary
to cause Diabetes Mellitus. He blocked the flow of pancreatic juice, removed
most of the pancreas, and grafted the small pancreatic remains just under the
skin of his test subjects, for easy removal at a later stage of the experiment.
The blood supply to this piece was left as normal as possible. At this stage no
diabetes was established. After removing the graft, Diabetes Mellitus could be
diagnosed immediately.
During the 1890s it was discovered that several diseases could be treated by
feeding patients extracts of thyroid. In analogy, it was tried extensively,
probably by more that 400 researchers worldwide [Bliss,
1982], to treat diabetic patients by feeding them pancreatic extracts,
without success. Mildly positive effects could never be reproduced by others.
Usually the toxic side-effects were far worse than the positive effects,
although sometimes the side-effects were seen as positive,
e.g. kidney failure could change the urine in such a way that
diabetes was no longer diagnosed.
In 1901 Opie showed a direct link between Diabetes Mellitus and damage to the
islets of Langerhans, generating a wide belief in an internal secretion in the
islets, responsible for the prevention of diabetes. In view of the failed
pancreas therapy experiments, scientists suggested that the exocrine pancreatic
secretion might destroy the active component of the internal secretion. Two
distinct types of pancreas seemed obvious choices for attempts to create pure
internal secretion: that from foetuses, in which islets develop before acinar
cells, and that from certain types of fish, which have anatomically distinct
islet and acinar parts. There are no records to prove the first was tried in the
early years, the second was tried between 1902 and 1904, but was not promising.
Several scientists all over the world attempted to isolate and purify substances
from the pancreas that were supposed to cure Diabetes Mellitus. Meanwhile,
several diabetologists kept believing that a diet was the only good way to treat
diabetics.
1.2 Earliest treatment of Diabetes
Mellitus
Until the 1910s opium was the only widely used medicine in the treatment of
Diabetes Mellitus. However, this could only dull the patients' despair, but did
nothing to cure or treat. Further treatment consisted of more or less trendy
diets. In the late 1850s Piorry advised the use of extra sugar, to compensate
for the loss of sugar into the urine. This 'eating a lot to compensate' was
practised until the early 1900s. In Paris under German siege, in 1870,
Bouchardat noticed that rationing of food caused the disappearance of glycosuria
in diabetic patients, while exercise also seemed to have a positive effect. The
idea settled that maybe the body should be put under as little metabolic strain
as possible by limited eating.
At the time of the earliest tests of pancreatic extracts in the treatment of
Diabetes Mellitus, America had two leading diabetologists who did not believe in
pancreas therapy. They were Allen and Joslin. They both practised 'starvation
treatment' where the patients are undernourished for a certain amount of time.
They argued that apart from the carbohydrate metabolism the protein and fat
metabolisms in diabetic patients were also affected. By cutting down on food
until the patient's body was relieved of all metabolic strain, and then slowly
building up again until a reasonably healthy diet was achieved, many diabetics
could live years longer. Some patients, however, did not even tolerate the
minimum amount of food (the 'living diet'), and succumbed quickly.
1.3 Towards reliable pancreas therapy
As early as the 1890s Paulesco, a Romanian scientist in Paris, developed an
interest in the internal secretion of the pancreas. He regained his interest in
1916, when he did his first experiments with extracts in Bucharest. The First
World War and Austrian occupation prevented serious experiments until 1919, and
he published some successes with his 'pancréine' in 1920 and 1921.
In the early 1900s Zuelzer, in Berlin, developed a pancreatic extract he
named 'acomatol' with which he managed to bring back a 50 year old patient from
a coma in 1906. This extract, produced for the Schering company, was probably
very contaminated and produced many side-effects. It was tested in Minkowski's
clinic in 1909, where it was concluded that the positive and negative effects
were due to the same component. This caused Zuelzer's funding to be withdrawn,
and he stopped publishing. He persisted with his experiments, however, and
produced a new extract for Hoffman-La Roche, for which he never published the
extraction methods. With hindsight, this extract was probably much better than
the first, if the convulsions that were reported were hypoglycaemic reactions
(signs of a low blood-sugar level).
Many other researchers worked on the extraction of the internal secretion of
the pancreas, both in Great Britain and the United States of America. Some, like
Dewitt and Scott, used pancreatic duct-ligation to atrophy the acinar cells.
This would remove the juice that would, as they believed, have destroyed the
internal secretion. Others used alcohol (like Zuelzer) to remove the pancreatic
juice. Among these were Knowlton and Starling, and Murlin and Kramer. Most of
their results were irreproducible. Two Americans, Kleiner and Meltzer, did
produce promising results of a decline in blood-sugar in depancreatised dogs,
caused by administering extract of the dogs' own pancreases. Most of the control
experiments were satisfactory. This work stopped abruptly in 1919 because
Kleiner left the laboratory [Bliss,
1982].
There were several meetings among those scientists to discuss the prospects
of the research. Especially the intervention of a Scotsman, Macleod, who worked
in Ohio, and who had been in contact with the researchers in Britain and seen
their disappointing results, caused some of the above to stop their
experiments.
On October 31, 1920, a fairly inexperienced general surgeon in London near
Toronto (Canada), Banting, read an article on pancreatic duct-blockage. This
gave him the idea of duct-ligation in order to isolate the internal secretion of
the pancreas. This, as seen above, had been tried before, but the article did
not mention it and Banting did not know. This is what he wrote in his note-book
[Bliss,
1982]:
"Diabetus
Ligate pancreatic ducts of dog. Keep dogs alive till
acini degener-
ate leaving Islets.
Try to isolate the internal secretion
of these to relieve glycosurea"
On November 8 he managed to arrange a meeting with Macleod, who worked in
Toronto at that time. Although Macleod had discouraged several scientists in the
field of pancreatic extracts, Banting's enthusiasm caused him to offer a
laboratory and some animals over the next summer holiday period, and the help of
a student, Best, as research assistant. In March 1921 Banting decided to take
Macleod up on his offer, after which the first dogs were depancreatised on May
17. Macleod had advised to use Hédon's method of pancreatectomy leaving a small
piece grafted under the skin to be removed later.
The first pancreatic extract was prepared and tested on July 30, with
temporary success. The dog died a day later. The pancreas used had been removed
seven weeks previously and left to degenerate. Another dog, brought back from a
coma, also died within a day. In order to speed up, a full pancreatectomy was
tried successfully on August 3, after which Hédon's procedure was not used
again. Banting and Best named their preparation 'Isletin' in notes on the
experiments on the dog that had the first total pancreatectomy.
Testing urine was sometimes difficult, because the volume of it decreased
after injections of extract; in some cases urination ceased altogether. But
during the second decade of the twentieth century methods of blood-sugar testing
had been developed and improved, which were much more accurate than urine
sampling. These tests made diabetes research more efficient and reliable.
Another time-saving idea was tried on August 17: extract of fresh,
non-degenerated pancreas. Banting and Best failed to recognise the positive
results and persisted with their faulty hypothesis that degeneration of the
pancreas was necessary to obtain pure internal secretion. Boiling extract
rendered it inactive, exhausting the pancreas' external secretion with secretin
was too effective. Extracts prepared with secretin lowered blood-sugar quickly
but caused profound shock. More and more control experiments were carried out,
e.g. in vitro sugar-burning capacity and checking the
activity of mixtures with trypsin. Macleod recognised it was this thorough
testing that needed to be extended in order to make it impossible for critics
and pessimists to deny the positive effects. The experiments would involve
pre-injection blood tests, more frequent blood sampling after injection so as
not to miss the effect, and establishing that it was a real blood-sugar lowering
effect rather than dilution phenomena caused by the injections of reasonably
large amounts of extracts.
In the first half of September 1921 different ways of injecting were tested.
Rectal injections had no effect, while subsequent intravenous injection did
prove effective. On September 17 injections were given subcutaneously for the
first time, but the results were not satisfactory, and Banting and Best decided
it was not worth trying again until they had trypsin-free extracts. By the end
of September two more respected Toronto doctors/scientists, Starr and Henderson,
had become involved in attempts to keep the promising research going by
providing lab space and money.
Some time between October and December 1921 Best read a publication by
Paulesco from July 1921. The blood-sugar data quoted by Paulesco were so
different from the generally accepted values for hyperglycaemia (probably due to
different techniques used by Paulesco), that Best was not impressed and chose to
ignore the information in the article.
In their first paper, describing work done up to November 10, which was
published in February 1922, Banting
and Best concluded that, although they had "always observed a distinct
improvement in the clinical condition of diabetic dogs after administration of
extract of degenerated pancreas", it was still too early for clinical trials. By
then, they had started a so-called longevity experiment, keeping a
pancreatectomised dog alive for as long as possible.
A visiting biochemist, Collip, became actively involved in designing better
experiments. Banting discovered information about foetal pancreases, from which
active extracts could be prepared without ligation or degeneration because of
the relatively low content of acinar tissue; this meant that extract could now
easily be produced in abundance from fresh, whole foetal pancreas. Time had come
to try to capture "the active principle". New bacterial filtering methods
produced more sterile extracts. Subcutaneous injections became possible,
spreading action over a longer period, thus preventing shock. A new blood-sugar
test was introduced, probably by Collip.
On November 23, Banting was injected subcutaneously with 1 1/2 cc of
Berkefeld filtered extract. The group had become impatient, wanted to get into
clinical testing. This extract did not seem to have any harmful effect, but
blood-sugar was not measured.
One longevity experiment ended on December 2; the dog died after convulsions,
due to anaphylactic shock. In retrospect, it could have been hypoglycaemic
shock, since more extract seemed to make it worse. The next longevity experiment
started four days later, in which the extraction was performed with alcohol
instead of aqueous saline, which was easier to evaporate in order to concentrate
the extract. This lead to the idea that the active principle could be extracted
from adult pancreases with alcohol too. Adult pancreases were available more
cheaply than foetal pancreas. Although Banting and Best had started using
alcohol and adult pancreases before Collip joined them, his experience and
expertise were much needed assets in the group. He started using rabbits, and
discovered that even healthy animals had a blood-sugar lowering response to
extract. He also found that the residue and not the filtrate of a final
filtering step contained the really powerful active principle. On December 20,
1921, Joe Gilchrist received tested, potent extract by mouth, with no benefit
after a day. At that time it still was not firmly established that only
injections would work.
Apart from testing urine and blood for sugar, Collip started testing the
urine for ketones and measuring the liver's glycogen, which show whether liver
function could be restored with the extracts. He also discovered hypoglycaemic
shock, a state of apparent toxic reaction which could be relieved by
administering glucose solution.
Because Collip was a much more self-sufficient, experienced and thorough
researcher, his results were much better received than those produced by Banting
and Best. Also, more and more people became involved in the experiments. This
made Banting feel he had been overtaken, and that his idea had been taken out of
his hands. Relationships within the group deteriorated quickly, and even became
violent at times. Because of this animosity, Banting and Best decided to have a
first official clinical test on January 11, 1922, on Leonard Thompson. The
timing was wrong. It was still too early, and the extract did not perform as
well as expected.
Meanwhile, Collip discovered the active principle could be purified to a
certain extent by gradually precipitating other protein with increasing amounts
of alcohol. Only at around 90% alcohol the active principle would precipitate,
leaving it pure enough not to cause abscesses at injection sites. His extract
underwent a first clinical test on January 23, also on Leonard Thompson. Two
days later the group signed an agreement to work together as a group again
rather than trying to compete with each other.
During February more clinical tests were carried out, all with favourable
results. At the same time, Paulesco started clinical trials, independently and
without knowledge of the work in Toronto. In April, the Latin-rooted name
'insulin' was proposed. The name 'insuline' had been suggested for the
hypothetical internal pancreatic secretion twice before, in 1909
and again in 1916,
by two independent scientists, and unknown to the Toronto group.
On May 3, 1922, the discovery of insulin was officially announced to the
medical world by Macleod.
1.4 The insulin molecule
After the actual isolation of insulin in 1922, it took another six years for
Wintersteiner
to establish that insulin is a protein. And it was not until 1955 that the
primary structure of insulin was elucidated by Sanger and co-workers. An account
of this process can be found in the transcription of Sanger's
Nobel Prize lecture.
1.4.1 Finding the primary structure
Based on the knowledge of protein chemistry in general and the composition of
insulin in particular, by 1943 Sanger started investigating the sequence of
amino acids of insulin. Chibnall
and his colleagues had shown a high content of free a-amino groups, i.e. a relatively high number
of N-terminal residues, one of which had been determined by Jensen
and Evans (1935) to be phenylalanine. Until 1952 Sanger believed the
molecular weight of insulin to be 12,000. In that year, Harfenist
and Craig showed it to be around 6,000, using the method of partial
substitution by 1-fluoro-2,4-dinitrobenzene (FDNB), separation of the reaction
products and colorimetric analysis of the monosubstituted derivative for the
dinitrophenyl group.
For further study of the N-termini, Sanger developed the DNP-method [1945],
which was later also used for many other proteins. The result was the discovery
of two phenylalanine and two glycine termini, along with non-terminal e-DNP-lysine. The assumed four polypeptide chains were
thought to be linked by disulfide bridges due to the relative abundance of
cysteines [du
Vigneaud et al., 1939]. By oxidation with performic acid [Sanger,
1949], the crosslinks were broken, resulting in two fractions, A and B.
Fortuitously, the two types of amino acids that could have confounded this
experiment by reacting with performic acid, methionine and tryptophan, are not
present in insulin. Fraction A contained the smaller number (around 20) of
residues, only 12 unique ones, of which none were basic. Four of them were
cysteine. Fraction B had 30 residues, two of which were cysteine. It seemed
there was only one type of glycine chain and one type of phenylalanine chain,
which was confirmed in 1949.
Mild acid hydrolysis of the DNP-derivatives of the fractions made it possible to
study the N-terminal sequences, resulting in Phe-Val-Asp-Glu (fraction B) and
Gly-Ile-Val-Glu-Glu (fraction A).
This meant there were only two types of chain, and not four different ones,
so the 12,000 molecular weight insulin was built up of two identical halves. Or,
alternatively, the actual molecular weight was 6,000. During 1950 Sanger
and Tuppy managed to sequence the whole of fraction B. Separation of the
complex partial hydrolyzate by various means resulted in simpler mixtures of
which direct analysis was possible. They ended up with a puzzle of about 45
peptides of varying lengths from which 5 sequences could be deduced:
| Phe-Val-Asp-Glu-His-Leu-CySO3H-Gly (N-terminal sequence)
|
(1) |
| Gly-Glu-Arg-Gly |
(2) |
| Thr-Pro-Lys-Ala |
(3) |
| Tyr-Leu-Val-CySO3H-Gly |
(4) |
| Ser-His-Leu-Val-Glu-Ala |
(5) |
Four amino acids were still missing, and it was impossible to establish how
the sequences would be joined together. The use of proteolytic enzymes (pepsin,
trypsin and chymotrypsin) [Sanger
and Tuppy, 1951] as hydrolytic agents instead of acid, solved this problem
by producing different peptides, which also included the missing residues.
Determination of the sequence of fraction A was completed by 1953.
This was more difficult because of the specific amino acid content and the lower
susceptibility to enzymatic hydrolysis. Ionophoresis at around pH 3 was needed
to separate the problematic cysteic acid peptides.
By that time, it was firmly established that the molecular weight of insulin
was, in fact, 6,000. So the only remaining question, that of the number and
nature of the disulfide bridges, was reduced to finding two between chains A and
B, and one intrachain bridge in chain A. In order to do this, unoxidised insulin
was subjected to hydrolysis to isolate peptides with intact cystines. Acid
hydrolysis was not suitable because of disulfide rearrangement reactions, but
enzymatic, neutral and alkaline hydrolysis all proved useful. Thus the complete
sequence of insulin was deduced (see figure
1.1).
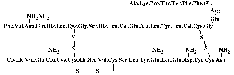
Figure 1.1: Sequence of insulin.
Sanger used bovine insulin
in his studies. Note the specific way of depicting amides. During strong acid
hydrolysis the amide groups of some glutamines and asparagines are also
hydrolysed. The real identity of these residues could be more appropriately
determined by enzymatic hydrolysis [Sanger
et al., 1955].
Human insulin differs from bovine insulin at positions A8, A10 and B30, where
it has threonine, isoleucine and threonine, respectively. The molecular weight
of human insulin is 5,808 [Hansen
and Brange, 1987]. The net charge of insulin is zero at pH 5.5, in good
agreement with the isoelectric pH of 5.3-5.35 as originally determined by Wintersteiner
and Abramson in 1933.
1.4.2 The three-dimensional structure of
insulin
Insulin had been crystallised for the first time by Abel
(1926). Over the years, the crystallisation method for these rhombohedral
crystals was standardised. The discovery of Scott
(1934) of the need for zinc in the crystals, was a big step forward in this.
In 1935 Crowfoot received a first sample of finely crystalline insulin. She
recrystallised the material according to Scott's method and took the first X-ray
photographs, which were published in the same year [Crowfoot].
They were not the first X-ray photographs of protein crystals ever, for pepsin
had been used before [Bernal
and Crowfoot, 1934]. The first crystallisations were approached as an
organic chemist would, so the crystals were dried with alcohol. Soon, however,
the positive influence of mother liquor was an established fact, and X-ray
measurements of wet insulin crystals were published [Crowfoot
and Riley, 1939].
The interpretation of the X-ray patterns had started with the use of Patterson's
ideas on the determination of the components of interatomic distances in
crystals. With the simplifications introduced by calculating Harker
sections only, the maps showed, in the case of insulin, strong features at
10 and 22 Å interatomic distances. When the first amino acid crystal
structure, glycine, was solved in 1939
through the application of the Patterson synthesis, solving protein structures
with the same methods still seemed "madness", according to Patterson himself, as
mentioned by Crowfoot
Hodgkin in 1968. The idea of replacing zinc isomorphously with heavier ions
had taken root almost as soon as the first photos had been taken. However, the
intensity changes introduced by cadmium were too small to be of any help.
Iodination experiments were not followed through, probably partly because of the
war, and because of the lack of experience with protein structure analysis in
general. Until around 1950 most of the interpretation consisted in building
models, almost by guessing and then checking whether they could produce the
diffraction patterns observed. All the proposed models exhibited the correct
symmetry, namely 32 [Hodgkin
and Riley, 1968], but nobody recognised the possibility of calculating the
correct size of the insulin molecule and the contents of the asymmetric unit
then. The idea of utilising multiple crystal forms in structure determination
was described by Crowfoot
in 1938. Unfortunately, the many different shapes of crystals observed all
turned out to be the same crystal form.
It was not until the primary structure of insulin became available in 1955
that Hodgkin's group in Oxford started devoting most of their time to the
problem of solving the three-dimensional structure of insulin. Schlichtkrull's
methods of crystallisation and his investigations into the exact amount of
zinc in those crystals, along with earlier experiments with cadmium insulin
crystallisation according to Scott, paved the way for the production of lead
insulin. It proved possible to remove zinc by soaking in EDTA and then replace
the zinc with lead. X-ray measurements were taken of the isomorphous series of
zinc-free, zinc, cadmium and lead insulin [Hodgkin
and Riley, 1968]. Especially the differences between metal-free insulin and
lead insulin were large, but the feasibility of solving the structure with those
alone, was still in doubt at that time.
However, it was not long at all until enough isomorphous heavy atom
derivatives were found to solve the structure to a resolution of 2.8Å. Five
derivatives were used for the phase determination:
- zinc-free insulin acetate, 0.01 M lead
- zinc insulin acetate, 0.1 M lead
- zinc insulin citrate, 0.02 M uranyl fluoride
- zinc insulin acetate, 0.01 M uranyl acetate
- zinc insulin citrate, mercuribenzaldehyde, saturated solution
Anomalous dispersion measurements were also taken. So late on Sunday night,
August 3, 1969, insulin was solved (for a facsimile of the original note
announcing the solution of the insulin structure, see Dodson,
Glusker and Sayre (1981)), and the structure of rhombohedral 2-zinc insulin
to a resolution of 2.8Å was published on November 1, 1969 [Adams
et al.]. The peptide chain, the disulfides and the aromatic
residues were well defined. A few regions, especially on the outside of the
molecule, were not completely clear.
After the determination of the primary structure of insulin by Sanger et
al. in 1955, several groups in China started the chemical synthesis of
insulin in 1958. They succeeded in 1965,
after which some pressure was applied to determine the three-dimensional
structure. The Cultural Revolution delayed the start until the beginning of 1967
[Tang,
1981]. The structure of insulin to 2.5Å resolution was published in 1971.
In 1972, when the Oxford group had extended their calculations to include data
to 1.9Å resolution, Hodgkin visited China and it was decided the two groups
would carry out further refinement separately and simultaneously. This resulted
in publications by the Beijing group of the structure at 1.8Å resolution
and at 1.2Å ,
and at 1.5Å resolution
by the Oxford group. Meanwhile, both groups started studying different insulin
species, for example insulin shortened at the B chain C-terminus [Peking
Insulin Structure Group, 1976], 4-zinc insulin [Bentley
et al., 1976], insulin crystallised in cubic form [Dodson
et al., 1978] and insulin from hagfish [Cutfield
et al., 1979].
1.4.3 Biosynthesis of insulin
The biosynthesis of insulin starts in the nucleus of the B-cells of the
pancreas. Most species have only a single insulin gene [Bell
et al., 1980]. The DNA contains two intervening sequences, one in
the 5' part of the mRNA that remains untranslated, and one almost in the middle
of the sequence for the C-peptide region of proinsulin, 179 and 786 base pairs
respectively. After transcription the mRNA ends up in the cytoplasm of the cell.
It is thought [Steiner,
1983] that translation of the mRNA into the 110 amino acid preproinsulin
(species: rat) starts while the ribosome is free in the cytosol. The signal
sequence of the preproinsulin anchors the ribosome to the membrane of the rough
endoplasmic reticulum (RER), after which the protein is translocated into the ER
lumen. The 59 "extra" amino acids in the preproinsulin appear to contain all the
information for accurate processing and secretion of the mature hormone. The
prepeptide has many hydrophobic side chains and is exactly long enough to span
the RER membrane so that the prohormone can start folding as soon as its
N-terminus enters the ER lumen. There is evidence that this anchoring function
of the prepeptide enhances the efficiency of protein folding. However, the
prepeptide will be cleaved off by signal peptidase on the inner surface of the
membrane before the whole of the peptide chain has entered the lumen. This is
necessary for the correct bridging of the last two cysteines, one of which is
the last-but-one in the nascent chain.
The length and not the amino acid content of the connecting peptide in
proinsulin (26-35 residues of greatly varying sequence for all known species)
appears its most important feature. Its function in folding and disulfide
formation, i.e. maintaining the correct spatial separation of B30
and A1, could just as easily be exerted by a much shorter peptide. The reason
for its excessive length may be that the prohormone chain needs to span the
distance from the interface between the small and large ribosomal subunits
(where translation takes place) to the inside of the ER, which is longer than 51
amino acids plus the necessary distance between B30 and A1 in the mature hormone
[Steiner,
1983]. This is an example of the 'minimum length hypothesis' [Steiner,
1981]. After folding and disulfide bridging, proinsulin is transferred to
the Golgi apparatus. There the proteolysis of the prohormone starts and the
mature hormone is concentrated, sorted and packed into secretory granules, ready
for extracellular release.
1.4.4 Insulin function
The actions of insulin have been known for quite some time [Steiner,
1977]:
- membrane transport of glucose, amino acids and certain ions;
- increased storage of glycogen;
- formation of triglycerides;
- stimulation of DNA, RNA and protein synthesis.
Three other peptide hormones are produced in the islets of Langerhans in the
pancreas:
- glucagon, consisting of 29 amino acids, in the A cells;
- somatostatin, a cyclic 14 amino acid polypeptide, in the D cells;
- pancreatic polypeptide, 36 amino acids with an amide C terminus, in the PP
cells.
Glucagon antagonises most of insulin's actions, while stimulating insulin
secretion. Somatostatin inhibits the three other islet hormones and a range of
hormones from different origins. Pancreatic polypeptide inhibits pancreatic
secretion altogether [Johnston
et al., 1988].
Once in the blood, insulin controls glucose homeostasis by stimulating the
uptake of glucose into skeletal muscle and, to a lesser extent, into liver and
adipose tissue. In muscle and adipocytes this uptake is mediated by the
so-called insulin-sensitive glucose transporter GLUT-4, a process that is not
yet understood. Other processes in the regulation of glucose homeostasis are:
alterations in glycogen metabolism in muscle and liver and decreased
gluconeogenesis in the liver. The enzymes involved in the insulin-regulated
processes of glucose metabolism appear to be regulated by (de)phosphorylation of
serine and/or threonine residues [Lee
and Pilch, 1994].
All known actions of insulin are initiated at the plasma membrane by insulin
receptors responding to ligand binding. A schematic view of the insulin receptor
can be seen in figure
1.2.
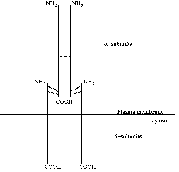
Figure 1.2: Schematic view of the insulin
receptor.
Reproduced from Lee
and Pilch (1994).
The a-subunit of 723 amino acids contains the site
or sites for insulin binding. The 620 amino acid b-subunit is built up of three regions: the extracellular,
transmembrane and cytosolic domains. Both subunits are glycosylated, resulting
in approximate molecular masses of 130 kDa and 95kDa respectively [Lee
and Pilch, 1994]. The insulin holoreceptor is composed of two a-subunits and two b-subunits
covalently linked by disulfide bridges to form a functional dimeric protein
complex. The receptor gene is translated into a single ab-product of which two are linked
together via disulfide bridges before being processed into separate
a- and b-subunits. The
covalent linkage of the two ab-heterodimers is unusual among the receptor/tyrosine kinase
family of which the insulin receptor is a member. The insulin holoreceptor can,
however, under mild conditions be reduced to ab-heterodimers which are functional monomeric receptor
species with a lower ligand binding affinity than the holoreceptor. The exact
position of the disulfide bridges both between a- and
b-subunits and between ab-heterodimers are not known; different studies are
contradictory [Lee
and Pilch, 1994].
Ligand-receptor contact occurs within the a-subunit.
The exact regions of contact remain incompletely defined despite a lot of
research effort. Various truncated versions of the receptor have been studied
for insulin binding and it seems that only those with an intact a-subunit are capable of binding the ligand, leading to the
conclusion that the entire a-subunit is important in
some way for insulin binding. At physiologically relevant hormone levels there
seems to be a stoichiometry of one insulin molecule per holoreceptor, with
negative cooperativity for binding of a second ligand [Lee
and Pilch, 1994]. Thus insulin somehow breaks the symmetry of the receptor
upon binding. Lee and Pilch propose a model for insulin binding to the receptor
(see figure
1.3a) where at physiological concentrations insulin makes contact
with two partial binding sites on separate halves of the receptor, resulting in
high-affinity binding. It can be seen from the model that at high concentrations
a second insulin molecule could potentially make contact with only one of the
partial binding sites, resulting in low-affinity binding (figure
1.3b).
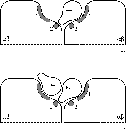
Figure 1.3: Schematic view of insulin binding as proposed by
Lee and Pilch.
a) One insulin molecule per holoreceptor at
physiological concentrations;
b) A second insulin molecule may bind,
less strongly, at high concentrations.
In essence, the model proposed by Lee and Pilch is the same as that proposed
by Schäffer
(1994), with high-affinity binding of the first insulin molecule to two
different partial binding sites on the two a-subunits
of the receptor, after which low-affinity binding of a second molecule is
possible to only one partial binding site. Schäffer implicates specific insulin
residues in the binding to both partial binding sites. The dimer-forming surface
of insulin (mainly residues B24, B25, A21 and B12, and potentially some residues
which are buried beneath the B chain C-terminus), named the 'classical binding
site', is best re-named 'binding site 1', and binds to receptor binding site 1.
It appears that the opposite side of the insulin molecule, known to be involved
in hexamer formation, forms 'binding site 2'. The two most important residues in
this site are leucines A13 and B17.
Following binding of the ligand, the receptor is rapidly autophosphorylated
on some or all of seven tyrosine residues in the cytosolic domain of the b-subunit. Exactly how the signal for autophosphorylation is
propagated from the site of ligand binding on the a-subunit to the intracellular autophosphorylation sites on
the b-subunit is unclear. The most likely explanation
seems a series of conformational changes, first between the a-a-subunits, then the transmembrane
domains of the b-subunits, followed by the b-b-subunits, after which
ATP-binding and autophosphorylation can take place [Lee
and Pilch, 1994].
The insulin receptor family primarily regulates nutritional metabolic
pathways, whereas all other receptor/tyrosine kinases mainly regulate cell
growth and differentiation. The same subdivision exists in that the insulin
receptor family has covalent links between the ab-heterodimers, and again in that these receptors do not form
direct complexes with substrates and/or effector molecules after
autophosphorylation. Instead, the insulin receptor, upon autophosphorylation of
at least the tri-tyrosine subdomain, acquires exogenous kinase activity,
phosphorylating its principal substrate: insulin receptor substrate 1 (IRS1).
IRS1, in turn, binds effector molecules which are responsible for the actual
processes of glucose transport and metabolism.
1.5 Research into modern treatment of
Diabetes Mellitus with insulin
Since 1922, a lot of research has gone into the improvement of insulin
therapy, both in terms of improved insulin preparations and ease of use.
Achieving purity was the first challenge. Abel
discovered insulin could be crystallised, which became standard procedure in
insulin purification only after Scott
(1934) established that zinc was needed in order to crystallise insulin in
rhombohedral form, a discovery inspired by his observation of zinc in the
pancreas. Another crystallisation step reduced allergic reactions [Jorpes,
1949]. Chromatographic techniques started to play a role in the 1960s,
leading to the first chromatographically purified insulin, Monocomponent insulin
[Schlichtkrull
et al., 1970].
The search for new insulin preparations with various desired properties was
approached in many different ways. At first it was thought the number of
injections needed every day could be reduced by using insulins with a retarded
uptake from the injection site, which could be achieved by introducing basic
additives as in protamine and isophane insulin [Hagedorn
et al., 1936 and Krayenbühl
and Rosenberg, 1946]. Addition of compounds like surfen or globin to acid
insulin solutions, which produce heavily insoluble complexes upon neutralisation
in tissue fluids, and complex formation of zinc with neutral insulin
suspensions, had the same effect. However, the need for strict metabolic control
in prevention of long-term complications called for the reinstatement of
multiple injections, along with the development of rapid-acting insulins for the
relief of the glucose-surge just after meal times, and mixtures of rapid-acting
and intermediate-to-long-acting insulins. Developments in this field include
Rapitard and Actrapid [Schlichtkrull,
1959 and Schlichtkrull
et al., 1961].
The duration of action could also be influenced by the physical state and
size of the insulin particles. Ultralente [Hallas-Möller,
1956] is an example of a long-acting crystalline insulin preparation.
Initially, only bovine insulin was used for crystalline preparations, having a
slightly longer action than porcine insulin in crystalline form. Gradually,
longer acting porcine preparations became available. Then, in 1979, recombinant
DNA techniques made it possible to produce human insulin in Escherichia
coli [Goeddel
et al., 1979]. Amazingly, this was done without knowledge of the
nucleotide sequence of the human insulin gene. Crea
et al. (1978) had chemically synthesised two separate genes
for chain A and B (77 and 104 base pairs for 21 and 30 amino acids,
respectively, plus start and stop codons and restriction site bases) following
nucleotide sequences that had been designed from the amino acid sequences. By
designing the nucleotide sequences the way they did, there was no need to
produce every possible trinucleotide separately. They produced 29
oligonucleotides, made from carefully chosen di-, tri- and tetranucleotide
building blocks. Goeddel
et al. (1979) describe the actual assembly of the genes, the
subsequent construction of the plasmid, and the expression and characterisation
of the product. They show that the amino acid content of the product is
indistinguishable from that of porcine insulin. It took another year before the
actual nucleotide sequence of the human insulin gene was published by Bell
and co-workers (1980). Only 21 out of 51 of the codons used by Crea et
al. turned out to be the same in the correct DNA-sequence.
Production of human insulin could also be achieved by conversion of porcine
insulin [Markussen,
1982] or biosynthesis in Saccharomyces cerevisiae rather than
E. coli [Markussen
et al., 1986]. Over the years, the production of many therapeutic
insulins has involved chemical alteration of the molecule. Nowadays, modified
insulins can be synthesised by mutation of the genes used in E. coli or
S. cerevisiae, thus facilitating structural and functional studies. But
apart from the scientific possibilities opened up by the availability of
recombinant insulin, there was a pressing need for a new source of the protein,
because the demand for insulin for therapy was outgrowing the supply of
slaughter pancreases for the isolation of insulin. Even now [Kott,
1996] the major insulin manufacturers are not able to provide insulin for
every area of the world, and cheaper beef and pork insulin is still produced,
especially for third world countries.
Because of the varying needs of diabetic patients, insulin has to be
available in multidose quantities. This requires the addition of antimicrobial
preservatives. Banting and Best used tricresol to that effect, and to this day
phenol and derivatives like m-cresol and methylparaben are used
throughout the range of therapeutic agents. Other additives include sodium
chloride or glycerol as isotonic agents, and certain buffers [Brange,
1987].
Alongside research into better and purer preparations, came the development
of techniques of administering them. At the time of Banting and Best it was
already established that oral therapy did not have any desired effect, and that
subcutaneous or intravenous injections were the only option. Many other routes
of absorption were tested, including rectal administration and absorption by
mucosae. Success was limited. Even with more modern technology of aerosol powder
[Wigley
et al., 1971], surfactants [Hirai
et al., 1981], liposome-enclosure [Dapergolas
and Gregoriadis, 1976] or polymer-crosslinking [Saffran
et al., 1986] the desired efficiency and bioreactivity has not been
achieved. The subcutaneous implantation of vinyl-ethylene copolymer pellets [Creque
et al., 1980 and Brown
et al., 1986] or biodegradable insulin-albumin microbeads [Goosen
et al., 1983], releasing insulin slowly and constantly over a
longer period of time, seems more promising. Most recently, reports have
appeared on glucose-responsive insulin release from certain polymeric systems
[Shiino
et al., 1995 and Valuev,
1995]. However, the only techniques in actual clinical use are based on
injections. Hospitals operate systems of continuous infusion, either according
to continuously measured glucose concentrations [Albisser
et al., 1974 and Pfeiffer
et al., 1974] or a pre-programmed schedule [Slama
et al., 1974]. These insulin pumps are not yet available to the
general public, although additional research is done on implantable pumps [Buchwald
et al., 1981]. Today's most patient-friendly portable insulin
delivery is the NovoPen, which has reduced injections to just pressing a button.
Most diabetics seem to prefer this to other available options [Jefferson
et al., 1985 and Walters
et al., 1985].
1.6 Classification of Diabetes
Mellitus
In 1979, the National
Diabetes Data Group formally classified Diabetes Mellitus and other
categories of glucose intolerance as follows:
- Type I, insulin-dependent Diabetes Mellitus.
Since this type usually
occurs in juveniles, it was previously called juvenile diabetes. It can,
however, start at any age. Patients usually present with easily recognisable
symptoms, so diagnosis is not difficult. Genetic determinants seem important
for the onset in most patients, with environmental factors a close second.
Abnormal immune responses (e.g. in normal childhood diseases
like mumps) and autoimmunity (see Bottazzo,
1993) are also thought to play an aetiologic role.
- Type II, noninsulin-dependent Diabetes Mellitus.
This type can become
recognisable at any age, although it can be asymptomatic for years and thus
usually presents in patients over 40 years of age. Often only the
complications seen after years of having diabetes, like neuropathy and
cataracts, cause a diagnosis to be made. Occasionally people are diagnosed as
a direct result of population studies, e.g. Mooy
et al., 1995 and Beks
et al., 1995, a study in Hoorn, The Netherlands. This research
into the prevalence of NIDDM and predictors for the development of the disease
resulted in 106 newly diagnosed diabetics out of 2,484 participants between
age 50 and 74. Not only age is an important factor in NIDDM. The genetic basis
of NIDDM seems even stronger than that of IDDM, and it is aggravated by
environmental factors. Moreover, 60 to 90% of all NIDDM patients in the
Western world are obese, which should be seen as an indicator for
subclassification of the type II diabetic. Symptoms are usually (at least
partly) alleviated by weight loss.
- Other types of diabetes.
Diabetes forms part of certain other
conditions and syndromes, whether obviously aetiologically related or not.
This class can be subdivided according to known or suspected aetiological
relationships, where diabetes may be secondary to
- Pancreatic disease (neonatal or later on in life);
- Hormonal abnormalities which may have either hypoinsulinaemia or
hyperinsulinaemia as a consequence;
- The administration of certain hormones, drugs and chemical agents, of
which oral contraceptives, tricyclic antidepressants and marijuana are but a
few;
- Insulin receptor abnormalities, either in the number of receptors or
their affinity for insulin, or even because of the presence of antibodies to
receptors (with or without associated immune disorders);
- Certain genetic syndromes, e.g. metabolism disorders,
insulin resistance, hereditary muscle disorders and cytogenic disorders like
Down's syndrome;
- Other types, of which diabetes associated with malnourished populations
is the most prominent example.
- Gestational Diabetes Mellitus.
There are two ways in which Diabetes
Mellitus and pregnancy can occur simultaneously. One is where a previously
diagnosed diabetic woman becomes pregnant, which involves certain risks for
both mother and child. The other is in Gestational Diabetes, when a pregnant
women becomes diabetic at some stage during pregnancy, because of the
pregnancy and most commonly only for the duration of the pregnancy. It is easy
to see the origin of problems in both cases, if one considers the fact that
even in a normal pregnancy the third trimester is a permanent state of mild
hypoglycaemia because all organs have to work harder than normal, some up to
50%. Sometimes the pancreas functions sub-optimally. This means that the
pregnant woman does not produce enough insulin herself, and the foetus will
start to produce insulin to attain an acceptable maternal insulin level. This
means the production of more foetal urine into the amniotic fluid. At the same
time the level of growth hormone in the foetus rises along with the foetal
insulin level [Remmers,
1994]. This causes macrosomia (birthweight >4,000 grams), which may
cause problems at birth. A Glucose Tolerance Test usually points out the need
for diet or insulin therapy. Oral hypoglycaemic treatment can not be used by a
pregnant diabetic [Drury,
1988]. Even after insulin treatment for GDM, most pregnant women return to
normal glucose tolerance after delivery. If this is not the case, they have
acquired clinical diabetes. From the previously gestational diabetics, 40%
acquire overt Diabetes Mellitus within 20 years [Coustan,
1993] so they should be kept under some sort of observation.
- Impaired Glucose Tolerance.
For the diagnosis of IGT an oral glucose
tolerance test is essential. The criteria for this classification lie between
those for normal subjects and diabetics. Consequently, the seriousness of the
disorder is intermediate between normal and diabetic, with some clinical
complications being completely absent while others, especially cardiovascular
abnormalities, commonly present. Thus, IGT may have prognostic implications
that should not be overlooked, especially in seemingly healthy individuals.
Like overt diabetes, IGT can be linked to numerous disorders and obesity.
Patients with IGT do not necessarily proceed to develop clinical diabetes;
many return to normal glucose tolerance for no apparent reason while others
stay in the IGT class for many years.
Apart from the clinical classes mentioned above, there are two so-called
statistical risk classes [NDDG,
1979]:
- Previous Abnormality of Glucose Tolerance.
This class is restricted to
those persons who have normal glucose tolerance but have previously
demonstrated diabetic hyperglycaemia or IGT either spontaneously or in
response to an identifiable stimulus. Re-classification of gestational
diabetics, former obese diabetics who have normal glucose tolerance after
weight loss, and temporarily hyperglycaemic patients (due to trauma or injury)
into this class is a useful tool for facilitation of follow-up of such
patients. The likelihood of such persons developing clinical diabetes (again)
should be considered to be increased.
- Potential Abnormality of Glucose Tolerance.
Persons who have never
exhibited abnormal glucose tolerance but who are at substantially increased
risk for the development of diabetes should be classified as PotAGT. Certain
risks for development of IDDM and NIDDM are well established, such as being a
relative of an IDDM or NIDDM diabetic or belonging to certain ethnic or racial
groups, although the degree of risk for any of the specific circumstances is
much less clear.
1.7 Studying the three-dimensional
structure of insulin
As described above, human insulin consists of 51 amino acids, divided into
two chains, commonly labelled A and B, with 21 and 30 amino acids respectively.
The chains are linked by three disulfide bridges, two forming interchain
cystines at A7-B7 and A20-B19, and one forming an intrachain cystine at A6-A11.
A piece of antiparallel b-sheet is formed upon
dimerisation: residues B23 to B28 of one monomer lie antiparallel to the same
stretch in the other monomer. There are two very small a-helices in the A chain, and a three turn a-helix running from residues B9 to B19 is found in every
insulin structure known so far.
Although insulin is only a small protein hormone with a characteristic
three-dimensional structure, the sequence varieties in nature and the biological
activities of these different forms in vitro, are quite diverse. This
diversity provides a tool for research into more effective and efficient
insulins for the treatment of Diabetes Mellitus. Solving modified insulin
structures will help understand the differences in structure-function relations
of all these insulins. It is also thought that understanding properties
(e.g. conformational changes) in a small protein like insulin will
contribute to the understanding of proteins in general.
File translated from TEX by TTH, version 1.54.
and edited manually
afterwards.
Back to the
Table of Contents

